


Fegato
Riduzione dei dotti biliari intraepatici (paucità dei dotti biliari) con possibile colestasi, ittero e prurito. Nei casi più gravi può essere indicato il trapianto di fegato.

Cuore
Stenosi e/o ipoplasia dell’arteria polmonare periferica, PPS (manifestazione più comune). Possibili altri tipi di cardiopatie congenite.
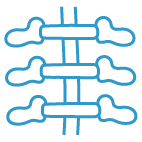
Scheletro
Vertebre ad ali di farfalla (manifestazione scheletrica più comune). Possibili altre lievi anomalie.

Occhio
Embriotoxon posteriore (manifestazione più comune). Descritte altre rare anomalie.

Fisionomia facciale
Viso a forma triangolare con una fronte ampia e prominente, occhi infossati, naso con punta arrotondata e mento piccolo e appuntito.

Sistema vascolare
Possibili anomalie intracraniche e sistemiche.

Reni
Possibili lievi anomalie strutturali e funzionali dei reni.